

17 June, 2019
Perovskite solar cells increasingly feature mixed‐halide mixed‐cation compounds (FA1−x−yMAxCsyPbI3−zBrz) as photovoltaic absorbers, as they enable easier processing and improved stability. Here, the underlying reasons for ease of processing are revealed. It is found that halide and cation engineering leads to a systematic widening of the anti‐solvent processing window for the fabrication of high‐quality films and efficient solar cells. This window widens from seconds, in the case of single cation/halide systems (e.g., MAPbI3, FAPbI3, and FAPbBr3), to several minutes for mixed systems. In situ X‐ray diffraction studies reveal that the processing window is closely related to the crystallization of the disordered sol–gel and to the number of crystalline byproducts; the processing window therefore depends directly on the precise cation/halide composition. Moreover, anti‐solvent dripping is shown to promote the desired perovskite phase with careful formulation. The processing window of perovskite solar cells, as defined by the latest time the anti‐solvent drip yields efficient solar cells, broadened with the increasing complexity of cation/halide content. This behavior is ascribed to kinetic stabilization of sol–gel state through cation/halide engineering. This provides guidelines for designing new formulations, aimed at formation of the perovskite phase, ultimately resulting in high‐efficiency perovskite solar cells produced with ease and with high reproducibility.
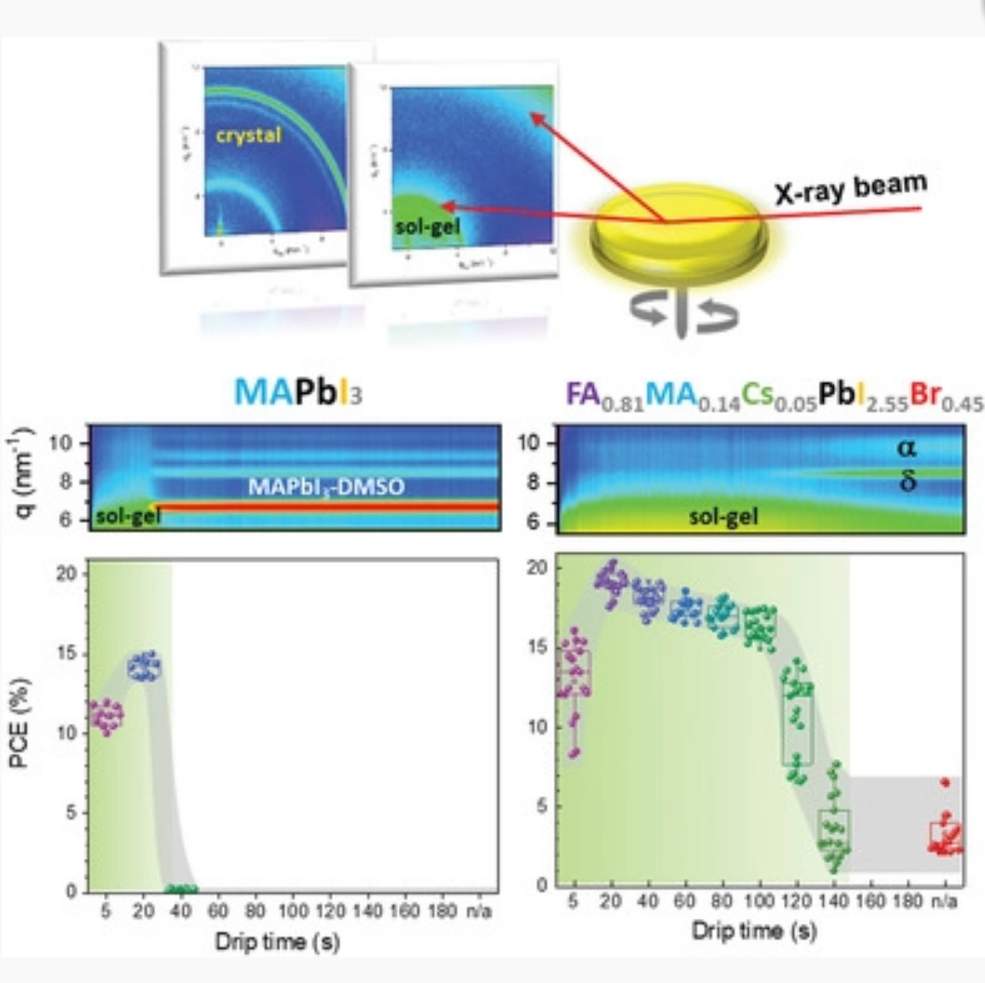
Figure. The role of cation and halide mixing is revealed using in situ X‐ray scattering measurements during spin‐coating. Modulating the cation/halide composition directly impacts the lifetime of the sol-gel precursor film and its easy and reproducible conversion to the perovskite phase to yield solar cells with 20% power conversion efficiency.
To read the full article please click.